Read & Write Data Functions
Compiled: May 11, 2026
Source:vignettes/articles/Read_and_Write_Functions.Rmd
Read_and_Write_Functions.RmdImport Data Functions
Seurat and other packages provide excellent tools for importing data however when importing large numbers of samples or samples with non-standard names this process can be cumbersome. scCustomize provides number of functions to simplify the process of importing many data sets at the same time and speed up the process using parallelization.
For this tutorial, I will be utilizing several publicly available data sets from NCBI GEO.
Import Single Data File
While most single cell packages have support for 10X and other commercial formats sometimes additional functions are required.
Import CellBender h5 File
CellBender is tool for the removal of ambient RNA (GitHub, Preprint). Starting with
CellBender v3 the output file is styled like Cell Ranger h5 files it
also contains some additional information. This causes
Seurat::Read10X_h5() to to fail when trying to import
data.
scCustomize has new function to read both new and old CellBender output files.
cell_bender_mat <- Read_CellBender_h5_Mat(file_name = "PATH/SampleA_out_filtered.h5")Import Multiple Data Files from Single Directory
Often when downloading files from NCBI GEO or other repos all of the
files are contained in single directory and contain non-standard file
names. However, functions like Seurat::Read10X() expect
non-prefixed files (i.e. Cell Ranger outputs).
scCustomize has three functions to deal with these situations without need for renaming files.
Import 10X Genomics (or 10X-styled) single directory with file prefixes
The function Read10X_GEO can be used to iteratively read
all sets of 10X style files within single directory.
For this example I will be utilizing data from Marsh et al., 2022 (Nature Neuroscience), which were downloaded from NCBI GEO GSE152183
list.files("assets/GSE152183_RAW_Marsh/")
GEO_10X <- Read10X_GEO(data_dir = "assets/GSE152183_RAW_Marsh/")
Read10X_GEO Additional Parameters
Read10X_GEO also contains several additional optional
parameters to streamline the import process.
-
parallelandnum_coresparameters enable use of multiple cores to speed up data import. -
sample_listBy defaultRead10X_GEOwill import all sets of files found within single directory. However, if only a subset of files is desired a vector of sample prefixes can be supplied tosample_list. -
sample_namesBy defaultRead10X_GEOnames each entry in the returned list (see below) using the file name prefix. If different names are desired they can be supplied tosample_names. - Several other parameters from
Seurat::Read10X(). See?Read10X_GEOfor more details.
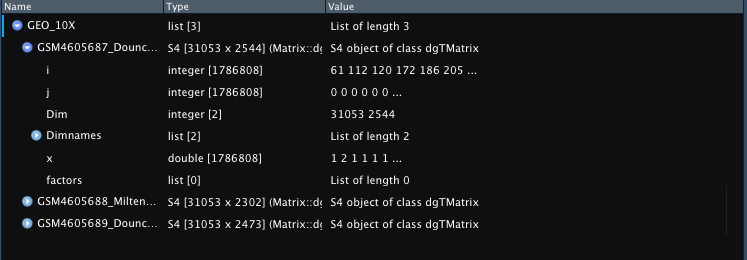
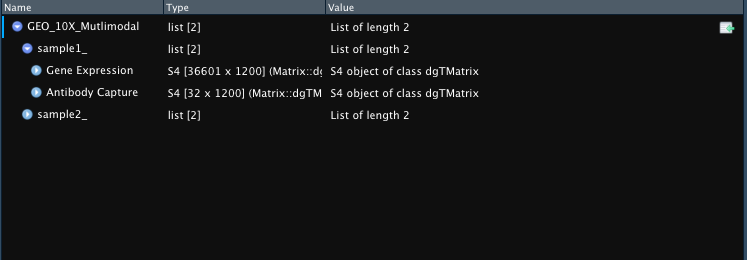 Example outputs for GEX only (list of matrices) or multimodal (list
of list of matrices) 10X-Style files downloaded from NCBI GEO
Example outputs for GEX only (list of matrices) or multimodal (list
of list of matrices) 10X-Style files downloaded from NCBI GEO
Import 10X Genomics H5 Formatted Files single directory with file prefixes
There is equivalent function for reading in 10X H5 formatted files
Read10X_h5_GEO.
NOTE: If files have shared aspect to file name specify this using
shared_suffix parameter to avoid that being incorporated
into names to list entries in returned list.
GEO_10X <- Read10X_h5_GEO(data_dir = "/path/to/data/", shared_suffix = "filtered_feature_bc_matrix")Import CellBender H5 Files from single directory
Importing CellBender h5 files from single directory can be done using
Read_CellBender_h5_Multi_File(), which functions very
similar to Read10X_h5_GEO. Here is example directory/file
setup:
Parent_Directory
├── Exp_Name
│ └── Cell_Bender_Results
│ └── SampleA_CB_out_filtered.h5
│ └── SampleB_CB_out_filtered.h5
│ └── SampleC_CB_out_filtered.h5
Multi_CB <- Read_CellBender_h5_Multi_File(data_dir = "Exp_Name/Cell_Bender_Results/", custom_name = "_CB_out_filtered.h5")Import delimited matrices single directory with file prefixes
Often data is uploaded to NCBI GEO or other repositories with single file (.csv, .tsv, .txt, etc) containing all of the information.
For this example I will be utilizing data from Hammond et al., 2019 (Immunity), which were downloaded from NCBI GEO GSE121654.
Read_GEO_Delim uses fread function for automatic
detection of file delimiter and fast read times and then converts
objects to sparse matrices to save memory
# Read in and use file names to name the list (default)
GEO_Single <- Read_GEO_Delim(data_dir = "assets/GSE121654_RAW_Hammond/GSE121654_RAW_Hammond/", file_suffix = ".dge.txt.gz")
# Read in and use new sample names to name the list
GEO_Single <- Read_GEO_Delim(data_dir = "assets/GSE121654_RAW_Hammond/GSE121654_RAW_Hammond/", file_suffix = ".dge.txt.gz",
sample_names = c("sample01", "sample02", "sample03", "sample04"))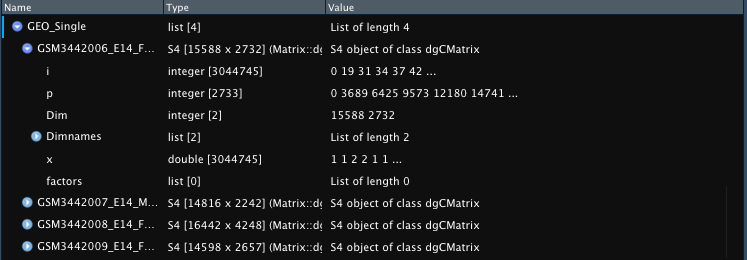
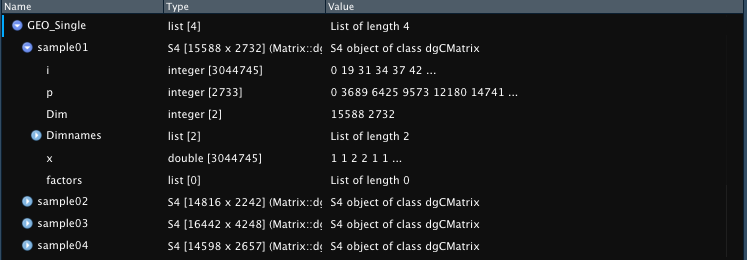 Example outputs with default naming based on file name or providing
Example outputs with default naming based on file name or providing
sample_names parameter.
Read_GEO_Delim additional parameters
See manual entry for more info.
Import Multiple Files from Multiple Directories
In addition to those functions for single directories, scCustomize
contains functions for when files are contained in multiple
sub-directories within shared parent directory.
NOTE: These functions all assume that each sub-directory contains
one sample and that sub-directory structure is identical between all
samples.
Import 10X Genomics (tsv, mtx)
Take an abbreviated example directory found below styled as output
from Cell Ranger count
Parent_Directory
├── sample_01
│ └── outs
│ └── filtered_feature_bc_matrix
│ └── feature.tsv.gz
│ └── barcodes.tsv.gz
│ └── matrix.mtx.gz
└── sample_02
└── outs
└── filtered_feature_bc_matrix
└── feature.tsv.gz
└── barcodes.tsv.gz
└── matrix.mtx.gz
# In this case we can use default_10X = TRUE to tell function where to find the matrix files
multi_10x <- Read10X_Multi_Directory(base_path = "Parent_Directory/", default_10X = TRUE)Path Inside Sub-Directories
In order to properly import the data
Read10X_Multi_Directory needs to know how to navigate the
sub-directory structure.
- The parameter
default_10Xtells the function that the directory structure matches the standardized output from Cell Ranger (see above). - However, it can also accept any unique sub-directory structure using
the
secondary_pathparameter as long as structure is the same for all samples (see below).
For instance:
Parent_Directory
├── sample_01
│ └── gex_matrices
│ └── feature.tsv.gz
│ └── barcodes.tsv.gz
│ └── matrix.mtx.gz
└── sample_02
└── gex_matrices
└── feature.tsv.gz
└── barcodes.tsv.gz
└── matrix.mtx.gz
# In this case we can use default_10X = FALSE to tell function where to find the matrix files
multi_10x <- Read10X_Multi_Directory(base_path = "Parent_Directory", default_10X = FALSE, secondary_path = "gex_matrices")Optional Parameters
Read10X_Multi_Directory also contains several additional
parameters.
-
parallelandnum_coresto use multiple core processing. -
sample_listBy defaultRead10X_Multi_Directorywill read in all sub-directories present in parent directory. However a subset can be specified by passing a vector of sample directory names. -
sample_namesAs with other functions by defaultRead10X_Multi_Directorywill use the sub-directory names within parent directory to name the output list entries. Alternate names for the list entries can be provided here if desired. These names will also be used to add cell prefixes ifmerge = TRUE(see below).
-
mergelogical (default FALSE). Whether to combine all samples into single sparse matrix and usingsample_namesto provide sample prefixes.
Import 10X Genomics (H5 Outputs)
scCustomize contains function:
Read10X_h5_Multi_Directory can be used to read 10X Genomics
H5 files similarly to Read10X_Multi_Directory
Import CellBender h5 Outputs
scCustomize also contains function
Read_CellBender_h5_Multi_Directory() which can be used to
read CellBender outputs in multiple sub-directories similar to
Read10X_h5_Multi_Directory using the same type of
parameters as Read_CellBender_h5_Multi_File
Merging Sparse Matrices
Rather than creating and merging Seurat objects it can sometimes be advantageous to simply combine the sparse matrices before creating Seurat object.
Basic Use
GEO_Single <- list(mat1, mat2, mat3)
GEO_Merged <- Merge_Sparse_Data_All(matrix_list = GEO_Single) Progress bar outputs in
Progress bar outputs in Merge_Sparse_Data_All function
through scCustomize.
Multimodal Data
If you have multimodal data (each entry in list contains sub-list
with matrices) then you can use
Merge_Sparse_Multimodal_All(). This function will return a
list with each entry representing a merged matrix for single
modality.
GEO_Merged_Multimodal <- Merge_Sparse_Multimodal_All(matrix_list = GEO_Multimodal)Add Barcode Prefix/Suffix
Merge_Sparse_Data_All contains a number of optional
parameters to control modification to the cell barcodes.
NOTE: If any of the barcodes in the input matrix list overlap and no
prefixes/suffixes are provided the function will error.
- Users can provide a vector of either prefixes or suffixes
to
add_cell_idsto ensure barcodes are unique (and make the import to Seurat smoother with samples already labeled).
- By default ids are added as barcode prefixes but can be added as
suffixes by setting
prefix = FALSE.
- The prefix/suffix delimiter is “_” by default but can be changed
using
cell_id_delimiterparameter.
Merge List of Seurat Objects
To easily merge many Seurat objects contained in a list scCustomize contains simple function.
# Merge a list of compatible Seurat objects of any length and add cell prefixes if desired
Seurat_Merged <- Merge_Seurat_List(list_seurat = list_of_objects, add.cell.ids = (c("cell", "prefixes",
"to", "add")))Write 10X Genomics Formatted H5 file from non-H5 input
Create_10X_H5 provides convenient wrapper around
write10xCounts() from DropletUtils package. Output can then
be easily read in using Seurat::Read10X_h5() or LIGER’s
createLiger() (which assumes H5 file is formatted as if
from Cell Ranger).
# Provide file path and specify type of files as either cell ranger triplicate files, matrix,
# or data.frame
Create_10X_H5(raw_data_file_path = "/path/matrix.mtx", source_type = "Matrix", save_file_path = "/path/",
save_name = "name")